DOI: ten.1039/C2BM00154C (Review Article) Biomater. Sci., 2013, 1, 347-360
Deoxyribonucleic acid origami technology for biomaterials applications
Received 20th October 2012 , Accustomed 12th November 2012
First published on 7th December 2012
Abstruse
Deoxyribonucleic acid origami is an emerging technology for designing and constructing divers multidimensional nanostructures. This engineering is at present expanding to materials science. This article introduces the basics of DNA origami, the design of various two-dimensional and iii-dimensional DNA origami structures, and the programmed assembly of origami structures. Deoxyribonucleic acid origami has unique backdrop, such as an addressable surface, which enables selective functionalization with biomolecules and nanomaterials. The origami can as well exist combined with top-down nanotechnology, such as placement on a fabricated substrate. The applied science is too practical to single-molecule imaging and assay systems constructed on designed DNA origami structures. Furthermore, DNA mechanical nanodevices working on Deoxyribonucleic acid origami have been realized, and cell-oriented applications are at present in progress. Dna origami engineering science has applied potential in diverse enquiry fields.
1. Introduction
Deoxyribonucleic acid materials science related to structural DNA nanotechnology has grown chop-chop as a inquiry area in the by two decades. The applied science allows the construction of various self-assembled scaffolds, which can be used for the placement and arrangement of functional molecules and nanomaterials and for the production of complex molecular devices. The field of DNA nanotechnology was pioneered by Ned Seeman, who created various important Deoxyribonucleic acid motifs and strategies for cocky-assembly which constitute the basic concept of structural Dna nanotechnology. one,2 DNA nanotechnology, which is now applied for the construction of nanoscale structures and functionalized materials, is further used in molecular mechanics and ciphering and in the fields of synthetic chemistry and biology, and information technology continues to develop in response to engineering demands. iii–five "DNA origami", a new programmed Dna assembly arrangement based on well-established DNA nanotechnology, enables the blueprint of two-dimensional (2nd) nanostructures with a wide variety of shapes of defined size. 6 Moreover, functional molecules and nanoparticles have been placed on DNA origami structures. 3–v
This article highlights the latest enquiry related to origami-based Dna nanotechnology and describes the expansion of the Deoxyribonucleic acid origami method to biomaterial applications. This review focuses on the basics of this technology, including the design and construction of diverse second and three-dimensional (3D) structures and programmed arrangements, and describes its application in the selective functionalization and single-molecule imaging of biomolecules, cell-targeted applications, and molecular machines built on a DNA origami scaffold.
ii. 2D Deoxyribonucleic acid origami
Dna origami, developed past Rothemund in 2006, has enabled the construction of a wide diversity of 2D structures of around 100 nm in size, including rectangles, triangles, and even a smiley confront and five-pointed star (Fig. ane). 6 In this method, a long single-stranded Deoxyribonucleic acid (M13mp18; 7249 nucleotides) and the sequence-designed complementary strands (chosen "staple strands"; most are 32-mer) are mixed so annealed from 95 °C to room temperature over two h, resulting in the formation of target structures past self-assembly (Fig. 1A). The construction tin can be imaged using diminutive force microscopy (AFM), and the assembled structure can be formed according to a design. To create 2nd Deoxyribonucleic acid origami structures, adjacent double-stranded DNAs (dsDNAs) should be connected to each other via a crossover. In this design, the geometry of the double helices involved has three helical rotations for 32 base of operations pairs (Fig. 1B). For instance, two neighboring crossovers of the central dsDNA in an arrangement of three adjacent dsDNAs should be located at the opposite sites (180° rotated, 0.5 turns). Therefore, the crossovers should exist separated past 16 base pairs (i.5 turns). To maintain a stable planar structure, this rule should be followed when placing multiple staple strands. DNA origami structures are formed using many unlike staple strands, so that hairpin DNA markers tin be placed at any position on the surface of the DNA structure. Hairpin DNA used every bit a topological marker is observed as a dot under AFM imaging. In this case, hairpins are placed perpendicular to the surface of the origami, therefore each hairpin should be placed at a position eight base of operations pairs from the crossover (270° rotation). The distance between the centers of the side by side staples is about half-dozen nm, so the side by side hairpins can be observed equally distinct spots according to the spatial resolution of the AFM. Using the hairpin markers, patterns, such equally the map of a hemisphere (Fig. 1D), can exist drawn precisely on the Dna origami surface.
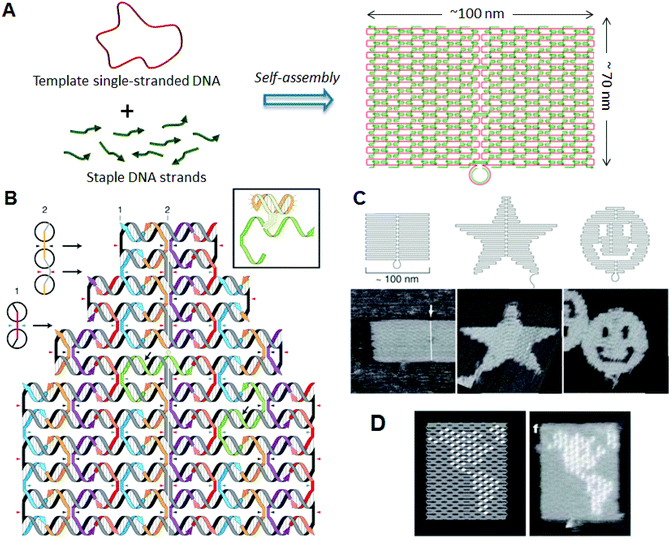 |
| | Fig. ane DNA origami structure. (A) The method employed to prepare a Deoxyribonucleic acid origami construction from the template unmarried-stranded DNA and staple strands. (B) Design of a self-assembled DNA origami construction and geometry of the incorporated dsDNAs. Colored strands and a greyness/black strand stand for staple strands and template unmarried-stranded DNA, respectively. Staple strands connect the next duplexes with crossovers. Inset: Structure of hairpin Deoxyribonucleic acid for a topological marker. (C) Design and AFM images of cocky-assembled DNA origami structures. (D) Drawing of a hemisphere on the DNA origami with hairpin DNAs (white dots) and an AFM paradigm of the assembled Deoxyribonucleic acid origami. | |
In addition, when functional molecules and nanoparticles are conjugated to specific staples, they can exist placed on the origami surface at selected sites. 2D structures formed with the DNA origami system non but have shape variations—a remarkable property of DNA origami is that all the positions of the construction have DNA sequence data (an address). Earlier the emergence of DNA origami technology, information technology was difficult to create ∼100 nm-sized structures through the self-assembly of small DNA components. Dna origami technology solved this problem and provided the breakthrough that enabled expansion of the shape design and the creation of addressable structures.
Since the DNA origami organization uses a long single-stranded template DNA, the size of the 2nd structure is determined by the length of the template strand. Various single-stranded DNAs were isolated for utilize as a template in the grooming of Deoxyribonucleic acid origami. 7,viii A strategy of using Deoxyribonucleic acid tiles (17 × xvi nm) instead of staples has also been developed, which allows size expansion past the introduction of 25 to 56 DNA tiles. 9
3. Programmed arrangement of multiple Deoxyribonucleic acid origami components
The programmed arrangement of DNA origami is an of import technique for preparing the desired big structures, peculiarly in terms of integrating complicated functions. Nosotros explored techniques for arranging multiple Deoxyribonucleic acid origami components, and developed methods to arrange rectangular DNA origami tiles horizontally in a programmed manner. ten As the ends of the helical axes align at both edges of the Deoxyribonucleic acid origami rectangles, a rectangular DNA origami tiles horizontally in a programmed fashion. ten In addition, every bit the ends of the helical axes align at both edges of the DNA origami rectangles, rectangular origami tiles assemble via π-interaction at the edges. Nosotros introduced specific concave and convex connectors into the origami rectangles to align these rectangles precisely with neighboring origami tiles. DNA rectangles should correctly assemble by shape and sequence complementarity, where complementary strands are introduced into the concavity and the convex connectors. Subsequently self-assembly, nosotros observed that the Dna tiles were aligned and oriented in the same direction. Furthermore, to align origami tiles accurately, the positions of the connectors and the concavity were changed to connect two specific tiles. V tiles were designed to marshal horizontally. In this organisation, we adopted a two-step self-assembly: offset, individual origami tiles were prepared, and so the multiple tiles were assembled in the second phase by slower annealing from 50 °C. The Dna origami is stable plenty to estrus at fifty °C in secondary annealing. To allow identification of the Dna tiles, hairpin markers were introduced onto the individual tiles. Afterwards self-assembly, judging by the social club of the markers, the five tiles were aligned correctly. In add-on, hairpin markers were used to display letters of the alphabet on the origami surface. The letters D, Due north, A, N, and O were each introduced onto one of the 5 tiles. After self-assembly using the showtime three tiles and last four tiles, the words "Deoxyribonucleic acid" and "NANO" were displayed, respectively. Using 5 tiles with the letters M, Y, O, T, and O, the five-letter word "KYOTO" was formed. We named these designed DNA tiles "Dna jigsaw pieces".
The method described above was practical to prepare the 2D assembly system. xi The shape and sequence selectivity were introduced to both lateral edges for extension in the vertical direction (Fig. 2A). 9 DNA tiles were designed and prepared. 3 tiles were so programmed to be connected vertically or horizontally, and three sets of vertical or horizontal trimers were finally assembled into a 3 × iii assembly with ∼30% yield; this assembly was confirmed by hairpin markers introduced onto the individual origami tiles. Taking a different approach, we explored novel 2nd assemblies. Four connexion sites on the four-way Dna origami connector were oriented to the outside to facilitate connectedness between the edges of neighboring DNA jigsaw tiles via π-interaction. 12 Using this iv-style connector, five and viii origami monomers were assembled to form a cruciform and a hollow square structure, respectively. Nosotros thus successfully created DNA origami-based second assembly systems. The method can exist extended to the construction of whatsoever construction past programmed self-assembly.
![Programmed self-assembly of DNA origami. (A) Structure of DNA origami having a concavity and a convex connector; the structure is called a]() |
| | Fig. 2 Programmed cocky-assembly of DNA origami. (A) Construction of DNA origami having a concavity and a convex connector; the construction is called a "DNA jigsaw piece" for 2D associates. A three × iii assembly of nine origami tiles and the AFM image of the associates. (B) Programmed assembly of multiple DNA origami structures using the assistance of scaffold frames. Target assemblies and their AFM images are shown. | |
Recently, Rothemund and co-worker created a programmed associates system by controlling the positions of adhesive π-stacking terminals for selective connection between rectangular tiles. thirteen They showed that a relaxed edge with blunt ends tin can form a stable connection, as opposed to a stressed border with the usual loop ends, which induce structural distortions. Multiple 2-dsDNA terminals with edgeless ends were introduced to gather complementary edges of the counterpart tiles as a binary code. In addition, the complementarity of the border shape worked effectively to precisely marshal the different tiles for the one-dimensional assemblies. The results indicate that the π-stacking interactions between the complementary edges can control the programmed assembly of multiple different origami tiles.
Yan and co-workers presented template-assisted assembly of Dna origami structures. xiv In this method, scaffold frames prepared from single-stranded template Dna and staple strands were used to straight the positioning of half-dozen–x predesigned Dna origami structures, including triangles, squares, and hexagons (Fig. 2B). By annealing the origami structures with connection strands and a scaffold frame, the target assemblies were obtained in a predesigned manner in relatively high yields.
The in a higher place strategy can be used to produce a larger assembly when practical to an origami assembly, and the positioning of the origami units tin can exist programmed using the defined design of the origami structures. The multifariousness of available 2D origami structures was expanded past the introduction of the predesigned and template-assisted strategies.
4. 3D Deoxyribonucleic acid origami structures
Given the geometry of the periodic double-helical Deoxyribonucleic acid construction, 3D structures can be designed by extending the second Dna origami system. Ii strategies for preparing 3D Dna origami structures accept been developed: one is the bundling of dsDNAs, where the relative positioning of adjacent dsDNAs is controlled by crossovers, and the other is the folding of 2D origami domains into 3D structures using interconnection strands. In the former method, developed past Shih and co-workers, relative positioning of adjacent dsDNAs is geometrically controlled by the crossovers. By arranging the positions of the crossovers, tubular and multilayered structures tin can be constructed (Fig. 3A). 15 Past increasing or decreasing the number of base pairs between crossovers (in this instance, two helical rotations for 21 base pairs), the relative positional relationship between adjacent dsDNAs is controlled. Using the rotational bending of 240° for seven base of operations pairs, three adjacent dsDNAs can be placed at a relative angle of ±120° with crossovers every 7 or xiv base pairs. Past alternate this relative positioning between adjacent dsDNAs, the duplexes course a pleated construction. When next dsDNAs are placed to rotate in i direction, the contiguous duplexes finally course a six-helix bundled tubular structure. Thus, when some parts of the pleated structures are turned backward by the introduction of i-directional rotation of next dsDNAs, the structures fold to become a stacked layer structure. In this case, to stabilize the 3D structures, next layers of dsDNAs should be farther connected past crossovers. Due to the complication and loftier density of the introduced crossovers, accurate folding into the target 3D structure requires a week-long folding fourth dimension. When the pleated structures were integrated as multilayered structures, the repeating units of the six-helix bundled tubular structures formed a honeycomb lattice, viewed from the centric direction of the double helices. It was also possible to create more than complex structures by perpendicularly joining these 3D structures. In addition, a wireframe icosahedron structure was assembled from iii double-triangle monomers made of a six-helix bundled tubular structure with connections. caDNAno software, which is publicly available, has been developed to support the design of these 3D structures. 16
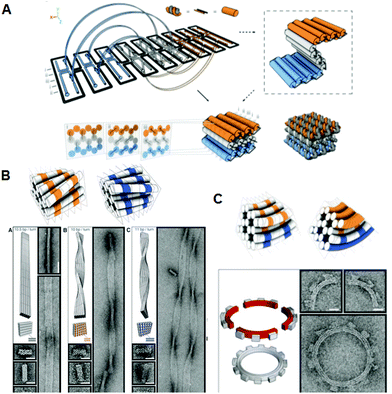 |
| | Fig. three Design and construction of 3D Deoxyribonucleic acid origami structures. (A) Scheme for folding the 2D pleated structure into a 3D multilayered structure using staple strands connecting adjacent layers. Sectional views of the positions of the crossovers in the multilayered structure sliced at vii-base-pair intervals. (B) Global twisted structures of six-helix DNA bundles obtained by the selective deletion or insertion of nucleotides to change the helical turns from the normal 10.five base pairs to 10 or xi base pairs. TEM images of the polymerized ribbons containing 10.five-base-pair, 10-base-pair, and eleven-base-pair helical pitches. (C) Global bending of six-helix Deoxyribonucleic acid bundles by the deletion and insertion of nucleotides in the adjacent duplexes. Assembly of 4 components of a quarter circle with 3 teeth (50 nm radius) and TEM images of the 12-tooth gear. | |
Furthermore, using the layered structures described above, new 3D structures were congenital by changing the helical twist from the average helical pitch of 10.five base pairs per turn to ten or xi base pairs per turn. 17 When dsDNAs having unlike helical pitches were arranged together, torque and repulsion between base pairs caused overall structural changes including twisting or thirty–180° bending (Fig. 3B). Using these structures as building blocks, left-handed or right-handed helical ribbon structures were prepared. In add-on, when angle-controlled duplex bundles were connected to each other, a vi-tooth gear and a spherical wireframe sheathing were created (Fig. 3C).
Using a unlike strategy, a Dna box construction was created by folding multiple 2D origami domains with interconnecting strands. 18 Vi independent rectangles were sequentially linked, and were designed to exist folded using interconnection strands in a programmed fashion (Fig. 4A, B). Analyses of the assembled structure by AFM, cryo-electron microscopy (cryo-EM), dynamic light scattering, and pocket-sized-bending 10-ray scattering indicated that the size was close to the original design. The hat of the box could be opened using a specific Dna strand to release the closing duplex past strand displacement, and the opening event was monitored past fluorescence resonance energy transfer (FRET) (Fig. 4C). Other types of DNA boxes accept been created using a similar method, which tin can command both the inside and exterior past adjusting the directions of the crossovers at the connexion edges. 19 A tetrahedral structure was designed and synthetic from four aligned origami triangles, which were preconnected with an M13 scaffold strand without folding independent 2nd plates. 20
 |
| | Fig. iv Design and construction of 3D structures from sequentially connected multiple rectangular plates. (A) Deoxyribonucleic acid box structure by folding of six Deoxyribonucleic acid origami rectangles using interconnection strands introduced at the edges of rectangles. (B) The DNA box model reconstructed from cryo-EM images. (C) Controlled opening of the box lid using selective DNA strands (fundamental). Lid opening result was monitored by FRET. | |
Using the strategy of folding 2d origami structures, we designed and prepared new hollow prism structures (Fig. 5A). 21 To make these structures, we offset prepared new Deoxyribonucleic acid origami structures having three, four, or six artillery, and and so introduced connexion strands to these multiarm Dna origami structures to allow them to be folded into 3D structures. After introducing the interconnection strands on the side edges of the artillery and annealing, no original 2D structures were observed, and linear, fiber-like structures appeared. The 3D structures constructed using this method were closed tubular and were characterized past AFM. Cryo-EM images revealed that the 3D structures were hollow prisms. Successive AFM scanning of the sample forced the closed structures to open into 2D structures (Fig. 5B). The tube-opening procedure was observed past high-speed AFM imaging, which could successively obtain one AFM prototype per second. The prism structures completely opened, with the number of scans necessary ranging from 1 to 30 depending on the individual prism. The opened prisms never reverted to their original multiarm structures. Later opening, the 2d structure formed a unmarried rectangle of ca. 130 nm × 90 nm, and the scaffold strand could be observed on the opened 2D construction.
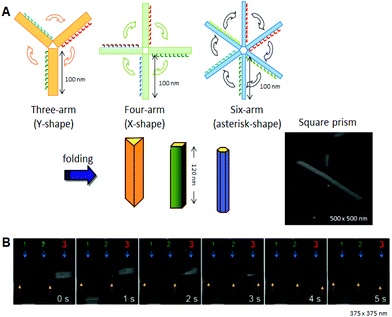 |
| | Fig. five Design and construction of prism structures and observation of their opening events. (A) Construction of various hollow prism structures by folding of multiple rectangular arms with connexion strands. The AFM image of the square prism structures. (B) The opening event of a prism (no. 3) visualized by successive high-speed AFM scanning (one epitome per second). | |
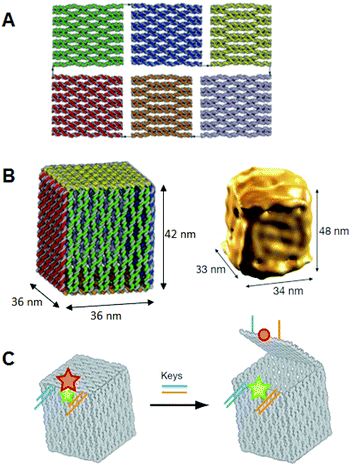
We further designed and constructed a cuboid structure using the design of a square prism structure. 22 Nosotros used the DNA origami method to set 6 rectangular DNA origami plates which were folded into 3D box structures using interconnection strands. 6 rectangular plates were designed, and the connection strands were introduced at the sides of the top and lesser plates to enable the prism construction to be closed into a box structure with a size of 36 nm × 36 nm × 44 nm. The six rectangles were formed subsequently annealing without connection strands. After annealing the staple strands with connectedness strands, the closed structures were observed by AFM. Dynamic lite scattering analysis showed the structure to be monodisperse (89%) and about 68 nm in diameter, which was like to the designed size (67 nm). The box-opening result was observed using high-speed AFM. Successive scanning of the sample converted the airtight 3D structure into 2D structures. The morphological changes occurred over 10 south, which was a detectable fourth dimension calibration for our AFM instrument. AFM also enabled u.s.a. to identify the individual plates involved in the opening process. The time scale for morphological changes of the 3D structures was tedious enough to be observed and characterized past high-speed AFM imaging. Dimensional conversion of ii dimensions into three dimensions could go a foundation technology for the manipulation of nanostructures. It is expected that target molecules will be caged in and released from the designed nanospace, and that chemical and enzymatic reactions inside the 3D structures will be realized.
5. Modification and functionalization of 2D Dna origami structures
5.1 Selective placement of functional groups
One of the about important features of DNA origami is that each private position on the 2D construction contains different sequence information. This means that the functional molecules and particles that are fastened to the staple strands can be placed at desired positions on the 2D structure.
By conjugating ligands and aptamers to staple strands, proteins were selectively attached on the 2D structures. 23–26 Combinations of specific proteins and ligands, such as SNAP-tag and halo-tag, were also used for the selective placement of fusion proteins on the DNA origami. 27
Yan and co-workers incorporated single-stranded DNAs to detect target RNA molecules on the DNA origami surface at the single-molecule level past AFM (Fig. 6). 28 Even though samples containing large amounts of cell-derived RNA were used, merely the binding of the target RNA could be visualized, and nonspecific binding was not observed. Once DNA origami tiles carrying different types of complementary DNAs were labeled with the corresponding hairpin DNA markers, binding of RNA targets could be identified from the specific hairpin markers on the Deoxyribonucleic acid origami, even though the different origami tiles were mixed. In this report, the detection limit of RNA molecules was virtually g molecules, meaning that the target RNA could be directly detected from a single cell without using polymerase chain reaction (PCR) distension.
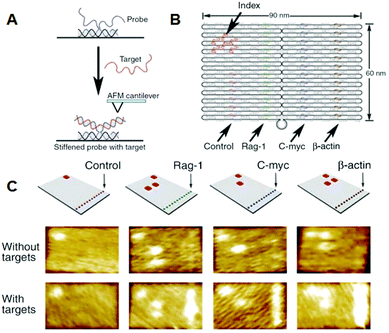 |
| | Fig. 6 Detection of target RNA by hybridization with probe DNA strands introduced on the Deoxyribonucleic acid origami. (A) The method for imaging the hybridization of target RNA to a probe DNA on the DNA origami. (B) Multiple DNA probes complementary to the target RNAs were introduced onto the Deoxyribonucleic acid origami and hairpin DNAs were also introduced equally an alphabetize for identifying the probe strand. (C) AFM images of bounden of target RNA to the probe strands. Specific DNA probes can be identified by the corresponding alphabetize. | |
Recently, functional molecules have been directly placed using sequence-selective ligands. Pyrrole-imidazole polyamide is a sequence-specific Dna recognition molecule, which binds to the target sequence in the minor groove according to a recognition rule. 29 We used a DNA origami structure with v cavities, into which v unlike sequences were incorporated. 30 The alkylation in the anchoring of the polyamide to the selective sequence and then the combined structure were visualized by streptavidin labeling using the conjugating polyamide with biotin. Employing this method, polyamide was found to alkylate the target sequence with 88% yield by discriminating a i-base mismatch. The selective alkylation and subsequent streptavidin labeling revealed the sequence selectivity of the polyamide at the single-molecule level.
Zn-finger protein is another sequence-selective molecule, and the binding sequence can exist selected by designing the amino acrid sequences. 31,32 Using DNA origami containing five cavities, nosotros introduced the strands with recognition sequences into each cavity. 33 The Zn-finger proteins bound direct to the crenel containing the target sequence with 50–eighty% yield. In add-on, green-fluorescence-protein-fused Zn-finger proteins were examined for bounden to the target sequences. The fusion of the poly peptide lowered the yield of the binding, however, the recognition ability was maintained. These results indicate that Zn-finger proteins tin be used as a carrier for positioning the target proteins.
These studies testify that the designed sequence-selective recognition molecules tin be used to identify the functional molecules and proteins on addressable Deoxyribonucleic acid origami structures.
5.ii Single-molecule chemic reactions
Selective bond cleavage and bond germination reactions were performed on a Deoxyribonucleic acid origami surface. Target organic molecules with specific reactivity were introduced into specific positions on the DNA origami. Reductive cleavage of disulfide bonds and oxidative cleavage of olefin by singlet oxygen were carried out on the DNA origami surface, and the reactions proceeded quantitatively at the single-molecule level. 34 In addition, amide bond formation and click reactions were performed with eighty–ninety% yield, and 3 successive reactions were also performed (Fig. 7). These chemical reactions were monitored past the cleavage of biotin-fastened chemical linkers and bond germination with biotin-tethered functional groups, which can be labeled with streptavidin for visualization by AFM.
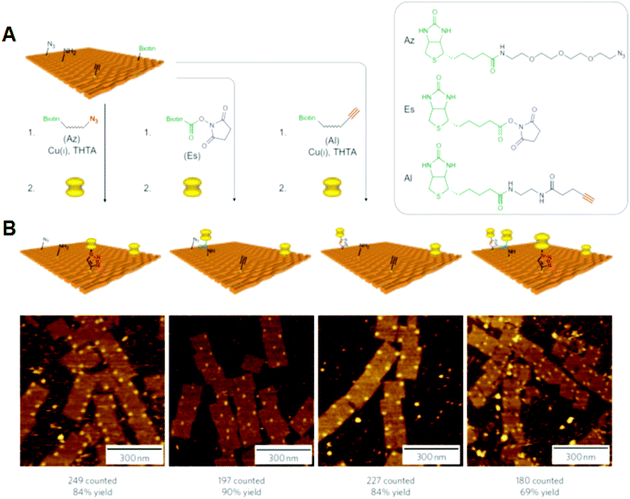 |
| | Fig. 7 Single chemical reaction on DNA origami. (A) Reactive groups (azido, amino, and alkyne groups) were incorporated into the DNA origami by conjugation with staple Deoxyribonucleic acid strands. The coupling reactions were then performed using the biotin-attached functional groups. The completion of the reactions was visualized by the binding of streptavidin. (B) AFM images of the 3 private reactions and three successive reactions by the treatment of three biotin-attached functional groups. Yields are presented below. | |
5.3 Selective incorporation of nanomaterials
The position-controlled placement of nanomaterials has been carried out by employing the addressable property of DNA origami. By directly coupling aureate nanoparticles (AuNPs) with a staple DNA strand, AuNPs have been selectively placed on Deoxyribonucleic acid origami. 35,36 Alternatively, thiol groups were placed on DNA origami structures to assemble AuNPs at predesigned positions. 37,38 The yield of the bounden of AuNPs improved when the small cavities of the origami structures were used to suit them. Incorporated single-stranded DNAs on Deoxyribonucleic acid origami controlled the positioning of two Dna-modified carbon nanotubes into cross-junctions on both sides of the DNA origami. 39 The position-controlled carbon nanotubes were practical in order to create a single-molecule device, which showed field-effect transistor-like behavior.
v.4 Placement of Deoxyribonucleic acid origami onto a fabricated solid surface
In another development, Deoxyribonucleic acid origami technology met superlative-down nanotechnology, including semiconductor processing. Fine triangular origami binding sites on the surface were made past electron axle lithography and dry etching. Origami binding sites on a SiO ii surface passivated with single-layer trimethylsilane (TMS), or origami binding sites on diamond-like carbon (DLC), were used for the alignment of triangular origami tiles. For the add-on of triangular origami, origami tiles were selectively aligned to the binding sites depending on the size of the sites (Fig. 8). 40 The hydrophilicity of the surface of the binding sites passivated with TMS is considered to be a driving strength for binding the Dna origami, whereas the interactions with binding sites on the DLC remained unclear. Using the various shapes of bounden sites on the surface, multiple origami triangles were attached selectively depending on the shape of the binding sites. Using a similar method, gold particles bound to the three vertices of a DNA triangle were successfully aligned to the origami binding sites with controlled orientation and arrangements. forty,41
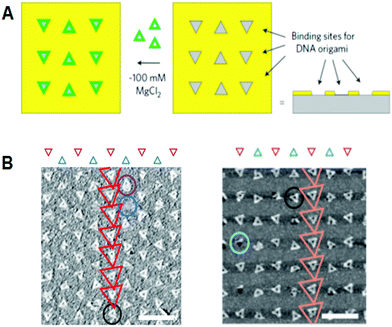 |
| | Fig. eight Alignment of DNA triangles onto the fabricated surfaces. (A) Fabricated triangular origami-binding sites were used for the alignment of DNA triangles. (B) Binding of Dna triangles onto the binding sites of TMS-layered SiO 2 surface (left) and DLC surface (right). Scale bar 500 nm. | |
Using the techniques developed in these studies, functional molecules and nanoparticles can be selectively placed at specific positions on the Deoxyribonucleic acid origami in a programmed fashion. The DNA origami arrangement can exist integrated with top-downwards nanotechnology. These methods can be applied to create nanoscale devices with novel functionality, when the functional origami is precisely integrated on the made surface.
6. Application to single biomolecule imaging
6.1 Control of DNA methylation and DNA repair in the DNA nanospace
Direct observation of enzymes interacting with DNA is expected to be i of the most significant technologies for investigating the mechanical behavior of enzymes. Considering DNA origami is used every bit a scaffold for AFM observation, the movement of biomolecules, including proteins and enzymes, and the Deoxyribonucleic acid structural change itself can be visualized and analyzed if the substrate dsDNAs and the target DNA structures are fastened onto the origami scaffold.
DNA modification using enzymes oft requires bending specific DNA strands to facilitate the reaction. The DNA methylation enzyme EcoRI methyltransferase (M.EcoRI) bends dsDNA by 55–59° during the methyl-transfer reaction. 42 To control the methyl-transfer reaction of Thou.EcoRI and examine the structural outcome on methylation, we designed and prepared a 2d Dna scaffold, named a "DNA frame", which accommodates two different lengths of dsDNA fragments: a tense 64-mer dsDNA and a relaxed 74-mer dsDNA (Fig. 9). 43 High-speed AFM revealed the unlike dynamic movements of the dsDNAs and complexes of Thou.EcoRI with 64-mer and 74-mer dsDNAs. Later on handling of the dsDNA in the Deoxyribonucleic acid frame with M.EcoRI and subsequent digestion by the restriction enzyme EcoRI, AFM analysis revealed that, compared with the 64-mer dsDNA, the 74-mer dsDNA was less finer cleaved, indicating that the methylation preferentially occurred in the relaxed 74-mer dsDNA, rather than in the tense 64-mer dsDNA. Biochemical analysis of the methylation and specific digestion using real-time PCR supported the higher up results. These results indicate the importance of structural flexibility in the angle of dsDNA during the methyl-transfer reaction with Grand.EcoRI. Therefore, Dna methylation tin can be regulated using the tension-controlled dsDNAs incorporated in the Dna frame nanostructure.
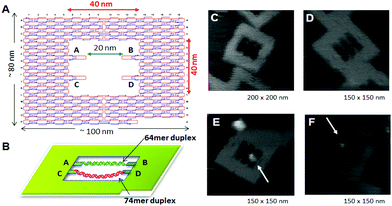 |
| | Fig. ix Control of the enzyme reactions in the DNA origami scaffold. (A), (B) Dna frame structure designed for incorporation of two different dsDNAs; tense 64-mer dsDNA and relaxed 74-mer dsDNA having the specific sequence for M. EcoRI at the center. AFM images of DNA frame (C), two-dsDNA fastened DNA frame (D), and M. EcoRI bound to the 64-mer dsDNA (E) and the 74-mer (F) dsDNA. | |
We next developed a novel method for the analysis of DNA repair by employing a Deoxyribonucleic acid frame containing various dsDNAs and using high-speed AFM. 38 Nosotros employed Dna base of operations excision repair enzymes, eight-oxoguanine glycosylase 44 and T4 pyrimidine dimer glycosylase, 45 for the analysis of a reaction on the defined DNA nanostructure. These enzymes accept glycosylase/AP lyase activity for removing damaged nucleobases and cleaving the Dna strand. 46 Nosotros placed various dsDNAs with a damaged base onto a Dna nanochip as a dsDNA cassette and analyzed the repair reaction at the single-molecule level. Nosotros placed 2 different lengths of substrate dsDNAs, tense 64-mer and relaxed 74-mer dsDNAs, onto a DNA frame to examine the structural effect on the glycosylase/AP lyase action, including cleavage of the DNA strand and trapping of reaction intermediates. The relaxed 74-mer dsDNA trapped the enzymes with NaBH iv reduction and was cleaved more than effectively compared with the 64-mer dsDNA. In add-on, dynamic move of the enzymes and the unmarried Deoxyribonucleic acid repair reaction were directly observed in the Deoxyribonucleic acid frame using a high-speed AFM. The DNA frame organisation serves to elucidate the detailed properties of the repair enzymes by direct observation of the events involved in DNA repair.
This method can be used for other Dna-modifying and repair enzymes that bend the double helix during the enzymatic reaction. The method can be extended to the directly observation of various enzymatic phenomena in the designed nanoscale space.
vi.2 Visualization of DNA structural changes
The germination and disruption of a single Grand-quadruplex structure were observed in a nanospace. 47 In that study, nosotros employed a Dna frame scaffold. To place the G-rich sequences, we prepared ii unique Dna strands (G-strands) which contained unmarried-stranded K-rich overhangs in the center for the formation of an interstrand Yard-quadruplex (Fig. 10). Three G-tracts were placed in the upper G-strand whereas the lower strand had a single Grand-tract. 48 The introduced strands were non in contact with each other. In the presence of 1000 + , the Grand-strands in the DNA frame conspicuously had an X-shaped structure, which indicated the formation of an interstrand G-quadruplex (Fig. 10, correct panel). The efficiency of the X-shaped formation was 44% using relaxed dsDNA. We further directly observed the dynamic formation of the 1000-quadruplex in real time by loftier-speed AFM. During scanning of the sample in the presence of M + , the 2 G-strands maintained a separated state for a given menses and then they suddenly formed an X shape. In a similar fashion, we observed the disruption of G-quadruplexes in the absence of K + . The X shape remained unchanged for a while, and and so reverted to the separated state under AFM scanning.
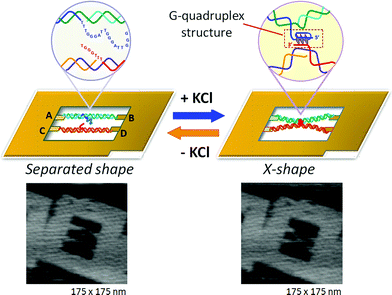 |
| | Fig. ten Visualization of Grand-quadruplex germination using the structural alter of two dsDNAs placed in the Deoxyribonucleic acid frame. In the presence of KCl, the separated state changes to the X-shape by connection at the center of two dsDNAs via Chiliad-quadruplex formation. | |
Nosotros thus realized the dynamic germination and disruption of Thou-quadruplexes by monitoring the structural changes of ii dsDNAs in the Dna nanostructure through loftier-speed AFM imaging. This was the first report of the existent-time ascertainment of a reversible conformational modify of a DNA construction. The method can be applied to the direct observation of diverse known conformational changes in nucleic acids.
seven. Application to DNA molecular machines
1 of the goals of the artificial molecular organisation is fully controllable motion of molecular machines. DNA molecular machines are operated past adding and removing specific DNA strands for complex movements. For this purpose, an actress sequence chosen a "toehold" is fastened to the end of the DNA strand. When a Deoxyribonucleic acid strand fully complementary to a toehold-containing strand is added, the initial strand containing the toehold is selectively removed at the toehold by strand displacement. The thermodynamic stabilization free energy works as "fuel" during hybridization to provide the mechanical movement of DNA machines. Using this strategy, Dna tweezers that perform shut–open up motions were constructed. 49 Besides created were two examples of a Dna walking device: a Dna walker with two legs that tin can control its direction of motion and a Dna motor that can movement frontwards autonomously by the cleavage of a DNA-nicking enzyme. fifty
Seeman and co-workers adult a controllable mechanical rotation device called a "PX–JX 2 device", which tin rotate two adjacent ends of dsDNAs by 180° in either the PX (paranemic crossover) or JX 2 (its topoisomer) state. 51–53 Two PX–JX 2 devices were introduced onto the origami scaffold at two specific sites to capture DNA nanostructures with four sticky ends through the ii sets of two complementary strands on the PX–JX 2 devices. 54 The PX and JX two states of the two devices allowed the binding of four different capture molecules with four unlike patterns of sticky ends corresponding to each rotational operation. Large DNA triangles were introduced onto the capture molecules as AFM markers for identification. Through the use of the four combinations of the PX and JX ii states, the specific capture molecules tin be trapped between the devices. In improver, 1 target capture molecule in a mixture of four different capture DNAs can correctly bind to the specific state of the device afterward heating to dissociate nonspecific capture molecules (fault correction).
Seeman and co-workers also examined the move of a two-legged Deoxyribonucleic acid walker forth tracks constructed in Dna nanostructures 55 and created a new DNA walker to operate the assembly line on DNA origami (Fig. 11A). 56 Three PX–JX 2 devices were fixed onto DNA origami, and the Dna walker moved along a pathway from the outside by following a predetermined operation. The movements of all the devices and the DNA walker were fully controlled by specific Dna strands. PX–JX two devices carrying AuNPs of various sizes and amounts could pass AuNPs to the walker by a rotational operation when the Dna walker was located nearby. The Dna walker moved in one direction along the runway. Three PX–JX 2 devices delivered AuNPs to the walker, and the DNA walker then picked upwardly the AuNPs at specific positions. The DNA walker, which somewhen picked up iii AuNP-spring DNAs, had a 43% yield. Every bit the on–off operation of the PX–JX 2 device delivering AuNPs was fully controlled past the specific Deoxyribonucleic acid strand, the final target product was obtained in a high yield (90%) with a very depression error rate (ane%). Moreover, using the on–off operation of three PX–JX 2 devices, eight patterns of AuNPs bound to the DNA walker were obtained.
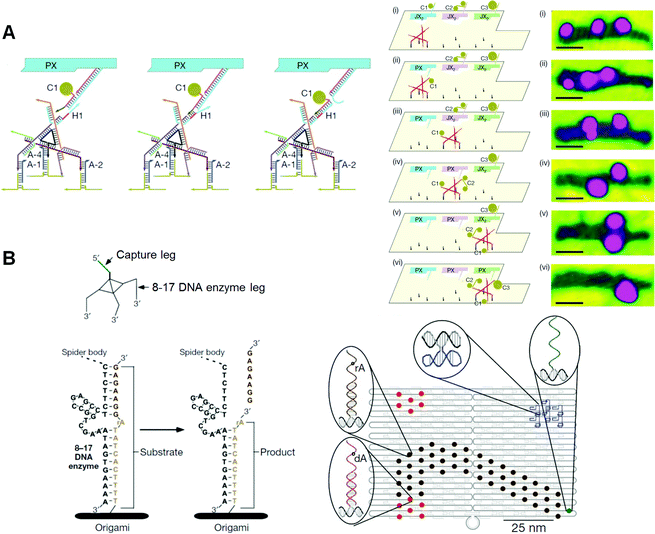 |
| | Fig. 11 DNA nanomachines walking on the DNA origami. (A) Assembly line of gold nanoparticle (AuNP) using a Dna walker. The DNA walker has four anxiety for walking on the selective position on the DNA origami and three easily to capture AuNPs. AuNP is transferred to the Dna walker from the PX–JX 2 device in the PX country past toehold-hybridization. The DNA walker stops at three specific places to capture AuNPs in PX state. Stepwise AFM images of AuNPs attached on the DNA walker after the operations. (B) DNA spider has three legs consisting of a DNAzyme to carve the RNA (rA)-containing Deoxyribonucleic acid strand introduced on the Dna origami. The Dna spider starts past releasing from the starting bespeak, which is placed with the capture leg, and and then walking on the substrate track past cleaving the substrates. The walker stops when information technology encounters the uncleavable substrate strands defective RNA (dA). | |
Stojanovic, Yan, and co-workers created a Deoxyribonucleic acid nanomachine called a "DNA spider", which has iii legs and one capture strand and moves along various patterned tracks constructed on DNA origami (Fig. 11B). 57 A DNAzyme that can hydrolyze RNA was incorporated into the iii legs of the Dna spider. Single-stranded DNA/RNA chimeras were incorporated into the DNA origami as a rail. A DNA spider was introduced and fixed at the starting position on the Deoxyribonucleic acid origami via a capture strand, and was so released from the starting bespeak using a specific Dna strand. The Dna spider was then spring to the Deoxyribonucleic acid/RNA chimeric strands in the track, and migrated forth the predetermined track by cleaving Deoxyribonucleic acid/RNA chimeric strands using DNAzymes in the legs. Finally, the spider stopped at a specific DNA strand, which did not comprise cleavable RNA. This study shows that offset, walk, and stop operations can be programmed into the predesigned track and DNA spider. The movements of the Dna spider were analyzed in real time by high-resolution total-internal-reflection microscopy, and the spiders were constitute to have moved beyond the Dna origami at three nm min −1 .
We have created a DNA transportation system in which a DNA nanomachine can motility along a designed track synthetic in DNA origami (Fig. 12). A track on a Deoxyribonucleic acid scaffold was constructed for observation of the multistep motion of a specific DNA strand. 58 Multiple ssDNAs (stators) were introduced as a track for hybridization of a complementary strand (motor strand). When the motor strand hybridizes to the specific stator, subsequent cleavage of the stator/motor duplex by a nicking enzyme Nt.BbvCI removes the short ssDNA of the stator, and the motor strand binds to the neighboring intact stator by branch migration and then finally steps forward. 59 We expected that multistep movement would be achieved by the turnover of the enzymatic reactions. Seventeen stators were introduced onto the Dna origami scaffold as a motor rail to observe the movements of the motor strand. The stator/motor duplex was introduced at site one of the motor track using a Dna scaffold defective the staple strand at site one. Subsequently annealing, a unmarried spot was observed at site 1, and the incorporation proceeded quantitatively. The DNA scaffold carrying the stator/motor at site 1 was then incubated for 0–3 h with Nt.BbvCI to examine migration of the motor strand along the Dna motor track. The motor strand was imaged equally a single spot of duplex on the DNA origami scaffold. We observed one-directional and fourth dimension-dependent movement of the motor strand forth the motor track. Furthermore, the movement of the motor strand forth the motor track was directly observed by high-speed AFM. The stator/motor duplex spot moved forrard along the motor track in each AFM paradigm with scanning every 5 south. In kymograph analysis, the distance of the motor-strand motion corresponded to the distance between next stators, indicating that the movement occurred stepwise on the track.
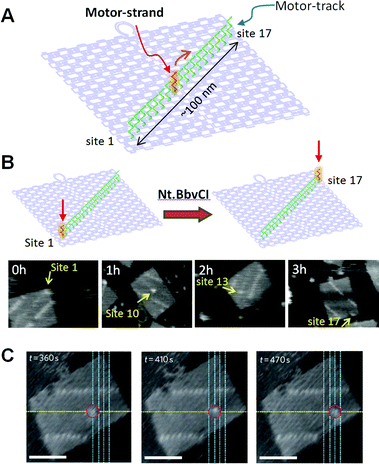 |
| | Fig. 12 Dna motor moving on the DNA origami. (A) A motor-rail (greenish ssDNAs) was constructed on the DNA origami and the movement of the Deoxyribonucleic acid motor (red ssDNA) was examined. (B) Time-dependent move of a DNA motor. (C) Stepwise movement of a DNA motor observed in existent-time past loftier-speed AFM. Scale bar 50 nm. | |
We further designed a complicated system for the control of the direction of the motor motility using a branched track and controllable blocking strands. 60 A branched track was constructed on Deoxyribonucleic acid origami, and three branching points and four concluding destinations were created. Blocking strands were introduced on both sides of the branching points to control the management of the Deoxyribonucleic acid motor. When ane side of the blocking strand was removed past the corresponding release strand, the motor strand could pass the branching bespeak in the predefined direction. The DNA passed ii branching points, therefore the ii releasing strands determined the pathway and destinations in a programmed fashion. The arrival at concluding destinations was observed by AFM and fluorescence quenching. The Dna motor found the predefined destinations by following the programmed instructions.
We thus designed and constructed a controllable Dna motor system using a track on a DNA origami scaffold. The stepwise movement of the motor strand was directly observed past loftier-speed AFM imaging. In addition, the direction of the movement was controlled past blocking and releasing of the specific strands. This method can exist applied to the development of a nanomachine that tin ship specific molecules and perform more complicated movements on expanded Dna nanostructures.
8. DNA nanostructures for cellar applications
The various applications of Deoxyribonucleic acid origami nanostructures described above have slap-up biological potential and have already been extended to cellular studies. A few examples of Deoxyribonucleic acid nanostructures being resistant to various types of endo- and exonucleases have been reported. 61 DNA origami constructs were able to maintain their integrity without degradation or damage in jail cell lysate of a series of cell lines. 62 The loftier stability of Dna nanostructures in a biological organization and their favorable compatibility with functional biomolecules, such equally proteins and aptamers, demonstrate that the nanostructures are promising biomaterials for living-cell analysis and as platforms for safe drug delivery. Unmethylated cytosine-phosphate-guanine (CpG) oligonucleotides with stiff immunostimulatory activities can exist recognized by endosomal Toll-like receptor 9 (TLR9) to induce immunostimulatory responses of the immune cells. 63,64 Multiple-branched DNA nanostructures and Dna tetrahedra begetting CpG motifs were developed for noninvasive intracellular deliveries. 65,66 Furthermore, taking advantage of a like triggering mechanism, Liedl and co-workers designed and constructed hollow tube-shaped origami which served equally an active carrier system for the investigation of allowed responses in mammalian cells. 67 The designs of the origami structure and the endocytotic pathway are illustrated in Fig. 13. Ane hollow Deoxyribonucleic acid origami structure (∼80 nm × 20 nm) with maximized surface area had as many as 62 inner or 62 outer binding sites (handle sequences, Hs) for CpG and anchor sequences (CpG Hs). The delivery performances and immunostimulatory responses of the Deoxyribonucleic acid tubes holding CpG were investigated in freshly isolated mouse splenocyte cells. The origami tube was first taken upward past immune cells, and then fused with a vesicle containing TLR9 segregated past a Golgi apparatus. The origami tube with CpG sequences was recognized by TLR9 in a vesicle, inducing the immune signaling cascade. Finally, a series of molecules, such as cytokines and CD69, were expressed to further stimulate the immune response.
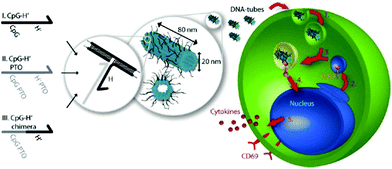 |
| | Fig. thirteen Schematic design of DNA origami tube and endocytotic pathway. Left: Three dissimilar types of CpG-H′ designed to hybridize with CpG. Right: endocytotic pathway of tube origami holding CpG with immune cells and after stimulating the immune responses. | |
Besides intracellular signal triggering, Douglas and co-workers created a hexagonal barrel chosen a Deoxyribonucleic acid nanorobot (shown in Fig. 14A) for transporting molecular payloads to target cells and subsequent multiple interactions with the cellular system. 68 Past employing an aptamer-based locking machinery, the reconfiguration of the nanorobot structure for payload delivery was triggered using sensing of molecules on the cell surface equally signal inputs. Aptamer-complement duplexes were introduced on the left and correct sides of the front end of the butt. This hexagonal barrel nanodevice can be unlocked in response to the protein keys between an aptamer-complement duplex and aptamer–target complex (Fig. 14B). The hollow inside tin be loaded with different types of payloads, such every bit proteins and nanoparticles (Fig. 14C, D), in a highly organized fashion for the delivery of various payloads, and the innate conformational regulation of the robot can be controlled selectively. To obtain a high yield of the nanorobot in a airtight state, two "guide" staples were incorporated shut to the locking site (Fig. 14E). The aptamer locking mechanism was designed to play the part of a logic gate by binding to the target cells. When the aforementioned aptamer sequences are used at two locking sites, the robot can be activated in response to but one blazon of key, whereas 2 types of inputs (cell surface antigens) as keys are required at the same time if two different aptamer sequences are employed in order to actuate the robot's role and expose the payload for farther interactions with target cells (Fig. 14F). Furthermore, this robot can be used to interface with cells and stimulate their signaling in an inhibition or activation manner by selective regulation of the nanorobot function. Therefore, DNA origami affords a new strategy for applications in cellular studies.
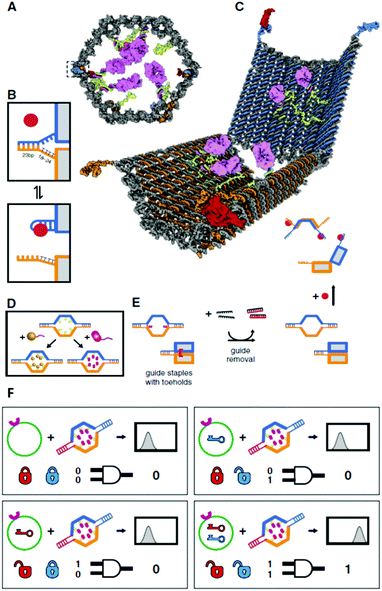 |
| | Fig. 14 Pattern of aptamer-gated DNA nanorobot. (A) Schematic drawings of the closed nanorobot in hexagonal barrel shape loaded with poly peptide payloads inside in front orthographic view. The nanodevice was fastened past two DNA aptamer based locks on the left (dashed box) and correct side. (B) The nanodevice is unlocked when a Deoxyribonucleic acid aptamer (orangish) and the complementary strand (orangish) dissociate in the existence of antigen central (red). (C) Schematic view of the open country of nanorobot by protein key displacement of aptamer locks. (D) Gold nanoparticles (gold) and Fab′ antibody fragments are employed every bit payloads inside of the nanorobot later on modifications. (E) Guide staples with toeholds are incorporated to obtain the loftier yield of nanorobot in the closed state. (F) AND-gated nanorobot is activated (aptamer-encoded unlock) past molecular inputs expressed by target cells. | |
9. Conclusion and prospects
A broad variety of designs of 2nd and 3D structures of effectually 100 nm in size take been realized by amalgam Dna origami. Compared with the employ of small Deoxyribonucleic acid assemblies, the Deoxyribonucleic acid origami method reduces experimental labor and the need for strict stoichiometry and eliminates uncertainties. In the relatively short fourth dimension since the DNA origami method was first reported in 2006, multidimensional structures, functionalization, unmarried-molecule imaging, and the structure of molecular machines take been realized. Functionalized Dna origami has already been combined with top-down nanotechnology, including semiconductor processing techniques. As the size of DNA origami is compatible with cellular uptake, it is expected that cell-targeting applications volition do good from the blueprint of various shapes and effective functionalizations.
It would be possible to utilize functional DNA origami equally a module to express college-level functionalities by assembling them in a programmed way. It is extremely difficult to adjust small-scale molecules in order to prepare desired structures. Nonetheless, if preassembled 100 nm-sized structures can be organized and purified to exclude incomplete structures, the usefulness of the structures will greatly improve.
Dna origami allows the precise placement and manipulation of functional molecules and biomolecules and is bachelor for the structure of a designed nanospace for chemical and biological reactions. Furthermore, equally the observation of the motion of biomolecules in a nanospace is at present possible, it is also possible to create devices that visualize the reaction and the behavior of biomolecules in the designed nanospace. This technology also opens the manner to expressing the circuitous functionality of the programmed arrangement of many different modules seen in living systems.
Notes and references
- N. C. Seeman, J. Theor. Biol., 1982, 99, 237–247 CrossRef CAS.
- N. C. Seeman, Nature, 2003, 421, 427–431 CrossRef.
- M. Endo and H. Sugiyama, Chem.–Eur. J. Chem. Biol., 2009, ten, 2420–2443 Search PubMed.
- A. Rajendran, Yard. Endo and H. Sugiyama, Angew. Chem., Int. Ed., 2012, 51, 874–890 CrossRef CAS.
- T. Torring, Due north. Five. Voigt, J. Nangreave, H. Yan and K. V. Gothelf, Chem. Soc. Rev., 2011, 40, 5636–5646 RSC.
- P. Westward. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS.
- B. Hogberg, T. Liedl and W. K. Shih, J. Am. Chem. Soc., 2009, 131, 9154–9155 CrossRef CAS.
- Eastward. Pound, J. R. Ashton, H. A. Becerril and A. T. Woolley, Nano Lett., 2009, 9, 4302–4305 CrossRef CAS.
- Z. Zhao, H. Yan and Y. Liu, Angew. Chem., Int. Ed., 2010, 49, 1414–1417 CAS.
- Thousand. Endo, T. Sugita, Y. Katsuda, Chiliad. Hidaka and H. Sugiyama, Chem.–Eur. J., 2010, 16, 5228 CrossRef.
- A. Rajendran, M. Endo, Y. Katsuda, Thousand. Hidaka and H. Sugiyama, ACS Nano, 2011, 5, 665–671 CrossRef CAS.
- M. Endo, T. Sugita, A. Rajendran, Y. Katsuda, T. Emura, Grand. Hidaka and H. Sugiyama, Chem. Commun., 2011, 47, 3213–3215 RSC.
- Southward. Woo and P. W. Rothemund, Nat. Chem., 2011, three, 620–627 CrossRef CAS.
- Z. Zhao, Y. Liu and H. Yan, Nano Lett., 2011, eleven, 2997–3002 CrossRef CAS.
- Due south. M. Douglas, H. Dietz, T. Liedl, B. Hogberg, F. Graf and Westward. Thou. Shih, Nature, 2009, 459, 414–418 CrossRef CAS.
- S. M. Douglas, A. H. Marblestone, S. Teerapittayanon, A. Vazquez, Thousand. M. Church and West. Thousand. Shih, Nucleic Acids Res., 2009, 37, 5001–5006 CrossRef CAS.
- H. Dietz, S. M. Douglas and Westward. M. Shih, Science, 2009, 325, 725–730 CrossRef CAS.
- E. South. Andersen, M. Dong, M. M. Nielsen, K. Jahn, R. Subramani, Due west. Mamdouh, M. M. Golas, B. Sander, H. Stark, C. L. Oliveira, J. S. Pedersen, Five. Birkedal, F. Besenbacher, Grand. V. Gothelf and J. Kjems, Nature, 2009, 459, 73–76 CrossRef CAS.
- A. Kuzuya and M. Komiyama, Chem. Commun., 2009, 4182–4184 RSC.
- Y. Ke, J. Sharma, Yard. Liu, Thousand. Jahn, Y. Liu and H. Yan, Nano Lett., 2009, 9, 2445–2447 CrossRef CAS.
- M. Endo, K. Hidaka, T. Kato, K. Namba and H. Sugiyama, J. Am. Chem. Soc., 2009, 131, 15570–15571 CrossRef CAS.
- M. Endo, K. Hidaka and H. Sugiyama, Org. Biomol. Chem., 2011, 9, 2075–2077 RSC.
- R. Chhabra, J. Sharma, Y. Ke, Y. Liu, Southward. Rinker, Due south. Lindsay and H. Yan, J. Am. Chem. Soc., 2007, 129, 10304–10305 CrossRef CAS.
- W. Shen, H. Zhong, D. Neff and Thousand. Fifty. Norton, J. Am. Chem. Soc., 2009, 131, 6660–6661 CrossRef CAS.
- Southward. Rinker, Y. Ke, Y. Liu, R. Chhabra and H. Yan, Nat. Nanotechnol., 2008, 3, 418–422 CrossRef CAS.
- A. Kuzuya, Yard. Kimura, K. Numajiri, N. Koshi, T. Ohnishi, F. Okada and M. Komiyama, ChemBioChem, 2009, x, 1811–1815 CrossRef CAS.
- B. Sacca, R. Meyer, M. Erkelenz, K. Kiko, A. Arndt, H. Schroeder, Yard. Due south. Rabe and C. M. Niemeyer, Angew. Chem., Int. Ed., 2010, 49, 9378–9383 CrossRef CAS.
- Y. Ke, Due south. Lindsay, Y. Chang, Y. Liu and H. Yan, Science, 2008, 319, 180–183 CrossRef CAS.
- T. Bando and H. Sugiyama, Acc. Chem. Res., 2006, 39, 935–944 CrossRef CAS.
- T. Yoshidome, M. Endo, G. Kashiwazaki, K. Hidaka, T. Bando and H. Sugiyama, J. Am. Chem. Soc., 2012, 134, 4654–4660 Search PubMed.
- J. G. Mandell and C. F. Barbas, third, Nucleic Acids Res., 2006, 34, W516–W523 CrossRef CAS.
- J. D. Sander, P. Zaback, J. K. Joung, D. F. Voytas and D. Dobbs, Nucleic Acids Res., 2007, 35, W599–W605 Search PubMed.
- E. Nakata, F. F. Liew, C. Uwatoko, S. Kiyonaka, Y. Mori, Y. Katsuda, M. Endo, H. Sugiyama and T. Morii, Angew. Chem., Int. Ed., 2012, 51, 2421–2424 Search PubMed.
- North. V. Voigt, T. Torring, A. Rotaru, Thou. F. Jacobsen, J. B. Ravnsbaek, R. Subramani, Due west. Mamdouh, J. Kjems, A. Mokhir, F. Besenbacher and M. Five. Gothelf, Nat. Nanotechnol., 2010, five, 200–203 CrossRef CAS.
- J. Sharma, R. Chhabra, C. S. Andersen, Grand. V. Gothelf, H. Yan and Y. Liu, J. Am. Chem. Soc., 2008, 130, 7820–7821 CrossRef CAS.
- B. Ding, Z. Deng, H. Yan, S. Cabrini, R. N. Zuckermann and J. Bokor, J. Am. Chem. Soc., 2010, 132, 3248–3249 CrossRef CAS.
- A. Kuzuya, N. Koshi, K. Kimura, K. Numajiri, T. Yamazaki, T. Ohnishi, F. Okada and Chiliad. Komiyama, Small, 2010, 6, 2664–2667 CrossRef CAS.
- M. Endo, Y. Yang, T. Emura, K. Hidaka and H. Sugiyama, Chem. Commun., 2011, 47, 10743–10745 RSC.
- H. T. Maune, S. P. Han, R. D. Barish, Chiliad. Bockrath, Westward. A. Iii, P. Westward. Rothemund and E. Winfree, Nat. Nanotechnol., 2010, 5, 61–66 CrossRef CAS.
- R. J. Kershner, Fifty. D. Bozano, C. Thou. Micheel, A. M. Hung, A. R. Fornof, J. N. Cha, C. T. Rettner, K. Bersani, J. Frommer, P. W. Rothemund and G. M. Wallraff, Nat. Nanotechnol., 2009, 4, 557–561 CrossRef CAS.
- A. M. Hung, C. M. Micheel, L. D. Bozano, L. Westward. Osterbur, G. M. Wallraff and J. N. Cha, Nat. Nanotechnol., 2010, 5, 121–126 CrossRef CAS.
- B. Youngblood and Northward. O. Reich, J. Biol. Chem., 2006, 281, 26821–26831 Search PubMed.
- M. Endo, Y. Katsuda, K. Hidaka and H. Sugiyama, J. Am. Chem. Soc., 2010, 132, 1592–1597 CrossRef CAS.
- Due south. D. Bruner, D. P. Norman and G. Fifty. Verdine, Nature, 2000, 403, 859–866 CrossRef CAS.
- One thousand. Morikawa, O. Matsumoto, M. Tsujimoto, K. Katayanagi, M. Ariyoshi, T. Doi, 1000. Ikehara, T. Inaoka and E. Ohtsuka, Science, 1992, 256, 523–526 Search PubMed.
- H. Chiliad. Nash, Southward. D. Bruner, O. D. Scharer, T. Kawate, T. A. Addona, E. Spooner, W. Southward. Lane and G. L. Verdine, Curr. Biol., 1996, 6, 968–980 Search PubMed.
- Y. Sannohe, M. Endo, Y. Katsuda, K. Hidaka and H. Sugiyama, J. Am. Chem. Soc., 2010, 132, 16311–16313 CrossRef CAS.
- Y. Xu, H. Sato, Y. Sannohe, M. Shinohara and H. Sugiyama, J. Am. Chem. Soc., 2008, 130, 16470–16471 CrossRef CAS.
- B. Yurke, A. J. Turberfield, A. P. Mills, Jr., F. C. Simmel and J. Fifty. Neumann, Nature, 2000, 406, 605–608 CrossRef CAS.
- J. Bath and A. J. Turberfield, Nat. Nanotechnol., 2007, 2, 275–284 CrossRef CAS.
- H. Yan, Ten. P. Zhang, Z. Y. Shen and North. C. Seeman, Nature, 2002, 415, 62–65 CrossRef CAS.
- S. P. Liao and N. C. Seeman, Science, 2004, 306, 2072–2074 CrossRef CAS.
- B. Ding and N. C. Seeman, Science, 2006, 314, 1583–1585 CrossRef CAS.
- H. Gu, J. Chao, South. J. Xiao and North. C. Seeman, Nat. Nanotechnol., 2009, 4, 245–248 CrossRef CAS.
- T. Omabegho, R. Sha and North. C. Seeman, Science, 2009, 324, 67–71 CrossRef CAS.
- H. Z. Gu, J. Chao, S. J. Xiao and N. C. Seeman, Nature, 2010, 465, 202–U286 CrossRef CAS.
- 1000. Lund, A. J. Manzo, N. Dabby, N. Michelotti, A. Johnson-Buck, J. Nangreave, South. Taylor, R. Pei, Yard. N. Stojanovic, Due north. Thou. Walter, East. Winfree and H. Yan, Nature, 2010, 465, 206–210 CrossRef CAS.
- Southward. F. Wickham, Chiliad. Endo, Y. Katsuda, K. Hidaka, J. Bathroom, H. Sugiyama and A. J. Turberfield, Nat. Nanotechnol., 2011, 6, 166–169 CrossRef CAS.
- J. Bath, Due south. J. Green and A. J. Turberfield, Angew. Chem., Int. Ed., 2005, 44, 4358–4361 CrossRef CAS.
- S. F. Wickham, J. Bath, Y. Katsuda, K. Endo, Grand. Hidaka, H. Sugiyama and A. J. Turberfield, Nat. Nanotechnol., 2012, 7, 169–173 Search PubMed.
- C. E. Castro, F. Kilchherr, D. N. Kim, E. L. Shiao, T. Wauer, P. Wortmann, M. Bathe and H. Dietz, Nat. Methods, 2011, viii, 221–229 CrossRef CAS.
- Q. Mei, 10. Wei, F. Su, Y. Liu, C. Youngbull, R. Johnson, S. Lindsay, H. Yan and D. Meldrum, Nano Lett., 2011, 11, 1477–1482 CrossRef CAS.
- J. Vollmer and A. M. Krieg, Adv. Drug Delivery Rev., 2009, 61, 195–204 CrossRef CAS.
- E. Latz, A. Verma, A. Visintin, One thousand. Gong, C. M. Sirois, D. C. Klein, B. G. Monks, C. J. McKnight, Grand. Due south. Lamphier, W. P. Duprex, T. Espevik and D. T. Golenbock, Nat. Immunol., 2007, 8, 772–779 Search PubMed.
- K. Mohri, 1000. Nishikawa, N. Takahashi, T. Shiomi, N. Matsuoka, G. Ogawa, 1000. Endo, K. Hidaka, H. Sugiyama, Y. Takahashi and Y. Takakura, ACS Nano, 2012, 6, 5931–5940 Search PubMed.
- J. Li, H. Pei, B. Zhu, L. Liang, M. Wei, Y. He, North. Chen, D. Li, Q. Huang and C. Fan, ACS Nano, 2011, v, 8783–8789 CrossRef CAS.
- Five. J. Schuller, Southward. Heidegger, N. Sandholzer, P. C. Nickels, N. A. Suhartha, Due south. Endres, C. Bourquin and T. Liedl, ACS Nano, 2011, v, 9696–9702 Search PubMed.
- South. G. Douglas, I. Bachelet and G. M. Church, Scientific discipline, 2012, 335, 831–834 CrossRef CAS.
|
| This journal is © The Purple Order of Chemistry 2013 |
0 Response to "3d drawing of structure of dna"
Post a Comment